
通过应对一系列不利条件,从DNA损伤到病毒感染,转录因子p53支持基因组稳定、细胞健康和生存。毫不奇怪,癌症谱系中的肿瘤携带p53突变,错误表达该蛋白,或调节其活性失调。几种信号通路,其中许多包含致癌蛋白,聚集在p53上控制其稳定性和活性。我们在此提出保守的激酶/ atp酶RioK1作为上游因子,在DNA、RNA和蛋白质水平上决定p53的活性。它通过将作用于p53的调控事件整合到一个连贯的反应回路中来完成这一任务。我们还将讨论RIOK1过表达如何代表癌症灭活p53的另一种机制,以及靶向RIOK1如何根除由失调的RIOK1 -p53网络驱动的恶性肿瘤。
保守的激酶/ atp酶RioK1通过在DNA、RNA和蛋白质水平上调节核糖体的生物发生、细胞周期进程、基因表达、代谢和生理来促进细胞生长和增殖。
RioK1对营养可用性作出反应,也可能对DNA损伤、热、渗透和氧化应激作出反应。
RioK1通过一个包含无数癌基因的复杂信号网络控制p53的稳定性和活性,从而控制细胞的健康和存活。
在整个癌症谱系中观察到,RioK1水平升高与高肿瘤分级、癌症侵袭性和低患者生存率相关。
升高的RioK1水平会触发p53降解并导致对放射治疗的抗性,而RioK1的消耗则会稳定p53并使癌细胞对治疗敏感。
癌细胞是否使用RIOK1过表达作为灭活p53的策略?
到目前为止,对RioK1的研究很少。因此,什么是它的全球靶点和在细胞生物学中未知的作用,导致它在失调时引发肿瘤发生、侵袭和转移?
在细胞和小鼠模型中,基因耗尽RioK1导致由构成RioK1-p53网络的癌基因驱动的肿瘤根除。因此,靶向药物的RioK1能否代表一种有效的、广泛作用的癌症治疗方法?
选择性抑制RioK1的药物并不存在。鉴于RioK1是一种结构非典型的激酶/ atp酶,是否可以开发出高特异性的靶向配体?
肿瘤抑制转录因子p53促进遗传稳定性、细胞健康和生存,以应对无数的压力。这些包括DNA损伤、营养剥夺、缺氧、热休克、核糖体耗竭、内质网应激、病毒感染和致癌激活[1]。自1979年发现p53以来,许多蛋白和调控rna已被证明可促进或拮抗p53的表达、稳定性和活性[1]。其中包括转录因子c-Myc和NF-κB、激酶Akt、PI3K、mTOR、Aurora A和B、gtpase H/N/K-Ras、G3BP应激颗粒组装因子2 (G3BP2)、E3泛素连接酶MDM2、E3 summoylase TRIML2、长链非编码rna MILIP和LincROR,以及microRNA mir - 2024 -5p。它们的活动如何结合成一个连贯的信号通路,接收上述不同的信号,然后将它们传递给p53,目前尚不清楚。
Chen等[2]最近的一项研究表明,激酶/ atp酶RioK1促进G3BP2磷酸化。这一事件引发p53泛素化和降解,并引起放疗抵抗。这一观察结果使我们进一步探索RioK1是否可以更广泛地参与p53的调控。事实上,决定p53活性的大量蛋白质和过程依赖于RioK1的表达、活性和稳定性。因此,我们在这里展示了RioK1-p53反应网络(图1A),其中包括riok1控制的事件(蓝线),这些事件向p53发出信号(橙线)。一些后一种调节因子反过来通过反馈回路影响RioK1的表达/活性/稳定性。RIOK1, p53编码基因TP53,以及位于RIOK1 -p53网络核心的蛋白质“节点”,在整个癌症谱系中被发现具有高频率的扩增、过表达或突变[3,4]。随之而来的RioK1-p53反应网络失调可能最终导致p53失活、肿瘤发生和无效治疗。
图1:RioK1及其下游网络调控p53活性。
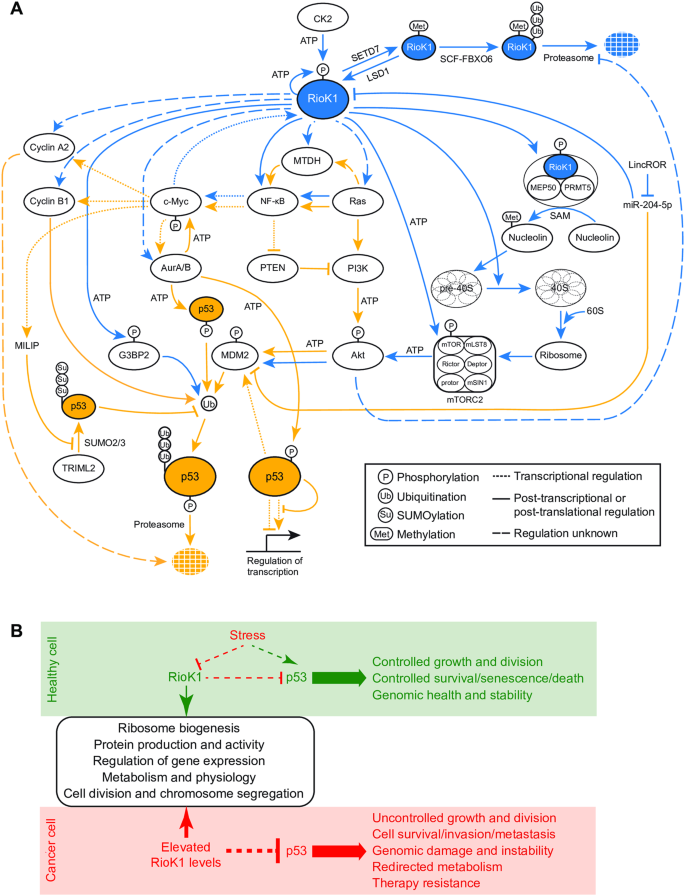
A .激酶/ atp酶RioK1的活性分别通过赖氨酸n -甲基转移酶SETD7、赖氨酸特异性去甲基化酶LSD1、酪蛋白激酶CK2和包含亚基FBXO6的E3泛素连接酶SCF复合物的甲基化、磷酸化和泛素化调节。如蓝线所示,RioK1促进40s前小核糖体亚基成熟,既通过释放生物发生因子,也通过作为适配器的一部分-作为蛋白质精氨酸甲基转移酶5复合体(PRMT5,也称为甲基化体)的一部分,激活核蛋白,调节pre-rRNA的产生和加工。RioK1也通过靶向mTOR激酶开启mTORC2激酶复合物。接下来,mTORC2激活蛋白激酶Akt,从而触发E3泛素连接酶MDM2复合物转化为多泛素化p53,从而使其被蛋白酶体降解。RioK1同样通过包括GTPases K/H/N-Ras和蛋白激酶PI3K的信号级联促进Akt活性。无论是直接还是通过Ras GTPases, RioK1支持metadherin (MTDH)的表达,MTDH通过NF-κB/PTEN/PI3K/Akt通路抑制p53。转录因子c-Myc驱动RioK1表达,并通过RioK1或RioK1- ras受NF-?B调控。c-Myc也激活激酶Aurora A和B的表达,后者磷酸化并稳定c-Myc。两种极光激酶都可以磷酸化p53的不同残基,从而降低p53的转录活性或刺激MDM2使p53多泛素化,随后发生蛋白酶体降解。c-Myc还激活长链非编码RNA MILIP的表达,MILIP通过竞争与p53结合,抑制TRIML2对p53的SUMOylation。后者通过阻止p53的多泛素化来保护它。长链非编码RNA LincROR像海绵一样,捕获并阻碍miR-204-5p活性,从而负向调节RioK1和MDM2。c-Myc进一步促进细胞周期蛋白A2和B1的表达,从而引起p53的降解。最后,RioK1通过直接激活G3BP2来下调p53, G3BP2通过MDM2刺激p53泛素化。橙色线表示调节p53稳定性、活性或转换的事件。B RioK1调控的过程示意图(圆角矩形)。在健康细胞中,RioK1-p53网络促进生长和活力;生存,衰老和死亡,并确保基因组的健康和稳定。在压力条件下,RioK1及其活性下调,而p53被激活。正如在癌症中观察到的那样,RioK1的过表达频率很高(图2A, C)会导致p53降解。后者导致遗传不稳定、生长和增殖失调、肿瘤发生、侵袭和转移以及对治疗的抵抗。详细信息请参见正文。
RioK1属于非典型蛋白激酶/ atp酶RIO家族,该家族还包括RioK2、RioK3和RioB,从古细菌进化到人类[5]。虽然RioK1是研究得最好的成员,但它已知的活性(图1A, B;表1)仍然有限。RioK1以参与核糖体的生物发生而闻名。作为PRMT5复合体(也称为甲基化体)的一部分,RioK1促进核蛋白的二甲基化,进而刺激rDNA转录、rRNA前成熟、rRNA折叠和核糖体组装[6]。作为一种atp酶,RioK1支持40s前小核糖体亚基成熟过程中生物发生因子的释放[7,8,9]。除了这些贡献,RioK1还磷酸化激活蛋白激酶Akt[10,11,12]。与RioK2一起,RioK1开启mTOR激酶,mTOR作为mTORC2复合物的一部分,磷酸化Akt[13]。因此,敲低RioK1会破坏核糖体产生和Akt信号传导,从而触发核糖体应激检查点[14]。后者激活p53(图1A),它停止细胞周期,然后通过凋亡消除被阻滞的细胞[13]。
当RioK1水平升高时,RioK1的靶点和活性最终导致p53降解。
耗尽RioK1还会影响关键细胞周期调节因子的磷酸化和蛋白水平,包括细胞周期蛋白A2和B1、激酶Aurora A和B、核糖核蛋白LARP1和微管不稳定蛋白stathmin-1[15]。这些观察结果强调了RioK1在控制细胞增殖中的重要性。RIOK1过表达通过上皮-间质转化(EMT)途径促进细胞迁移和侵袭。由于转录因子STAT3和TWIST1下调,E-cadherin水平升高,N-cadherin、vimentin和基质金属蛋白酶-2浓度降低,敲低RioK1会使EMT活性消失[2,11,15,16]。在相同条件下,促进Akt-和NF-κ b -介导的信号传导和转移的致癌蛋白metadherin (MTDH)表达减少[15](图1A)。
原致癌转录因子c-Myc与反激活子MAPJD一起驱动RIOK1表达[19],而miR-204-5p通过下调RIOK1转录物来拮抗c-Myc[10](图1A)。反过来,RioK1促进c-Myc mrna的翻译[20],从而建立一个反馈回路,刺激c-Myc的转化和转移能力。由于c-Myc和RioK1参与相同的过程(包括基因表达、核糖体生物发生、细胞周期控制、代谢、运动和侵袭),RioK1很可能是c-Myc介导的肿瘤发生的关键下游促进剂。RIOK1的表达也受到E2F转录因子的刺激[21]。其启动子还包括一个CpG岛,其染色质富含转录激活H3K4me3标记[21]。ChIP-Seq和ChIP-chip实验分别揭示了致癌转录因子FOXM1和肿瘤抑制赖氨酸特异性组蛋白去甲基化酶6a (KDM6A)在RIOK1启动子上的存在[22]。需要进一步的研究来破译RIOK1在表达水平上的调控,这似乎比目前所认识的要复杂得多。
RioK1的水平和活性也在翻译后受到控制(图1A)。赖氨酸n -甲基转移酶SETD7在K411位点甲基化RioK1,促进其与FBXO6的相互作用;E3泛素连接酶复合物SCF的亚基。后者泛素化RioK1以触发其蛋白酶体降解。相反,酪蛋白激酶2 (CK2)在T410位点磷酸化RioK1以防止K411甲基化,而赖氨酸特异性去甲基化酶1 (LSD1)通过SETD7逆转RioK1甲基化[9,16](图1A)。CK2在体外也能磷酸化S21和S22的RioK1[23]。RioK1在S407位点磷酸化自身[23],可能是为了防止其寡聚化并维持其最活跃的单体形式[24]。重要的是,在结直肠癌中,RIOK1与SETD7呈负相关表达,而RIOK1与LSD1或CK2呈正相关表达,进一步证实了RIOK1在恶性环境中的上调[16]。
与RioK1共纯化的蛋白质/底物的生物活性[25]表明该酶的作用超出了迄今已知的范围。这些包括应激反应、代谢、核糖体翻译活性、蛋白质周转、染色质重塑和转录调控、RNA加工和周转、着丝粒组装和活性。在酿酒酵母中,与同源物Rio1相互作用的蛋白质介导类似的功能[25,26],表明RioK1/Rio1具有许多尚未被探索的功能。例如,我们实验室最近的一项研究表明,Rio1和RioK1下调着丝粒转录物水平,以确保结构合适的着丝点及时形成,从而促进细胞分裂过程中染色体的忠实传递[26]。
摘要。
事实。
开放式的问题
介绍
RioK1:一种多用途激酶和atp酶
riok1调控的事件影响p53的稳定性和活性
RioK1作为广谱抗癌药物靶点
参考文献。
致谢。
作者信息
道德声明
# # # # #
如前所述,RioK1缺失的细胞触发核糖体应激检查点[13],该检查点向p53发出信号,停止细胞分裂并诱导细胞凋亡[13]。就在最近,RioK1被证明在T226位点磷酸化G3BP2蛋白,从而引发p53泛素化和降解[2](图1A)。这两个发现都强调了RioK1和p53之间的重要关系。仔细观察目前已知的RioK1蛋白靶点(图1A中的蓝线)和调节p53的蛋白质和rna(图1A中的橙线),使我们能够将它们联合到一个连贯的信号网络中。考虑到所涉及的蛋白质的作用,这个多功能网络作为应激反应、核糖体产生和活性、代谢、细胞生长和分裂、凋亡、肿瘤发生、迁移和侵袭的管理者出现(总结于图1B)。由于过表达、基因扩增或稳定性增强而导致的RioK1活性改变(图1A和图2A、B)可能与MDM2过表达类似,导致p53失活,导致无限制的生长和增殖、遗传不稳定、生存、侵袭和转移增加,以及无效的癌症治疗(图1B)。
图2:RIOK1 misex的频率在整个癌症谱系中出现的变异、放大或改变。
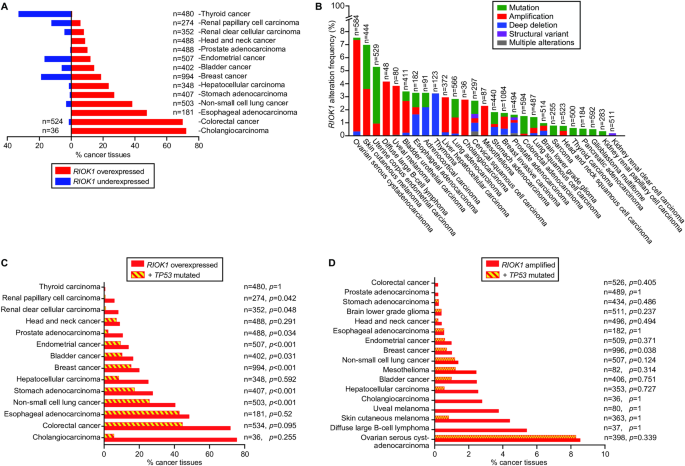
错误表达RIOK1的癌症百分比。N=病例数。B携带RIOK1特异性改变的癌症百分比。N=病例数。C仅过度表达RIOK1和携带TP53突变的癌症的百分比。n=病例数,p值表示RIOK1过表达和TP53突变在同一癌症中发生的统计可能性。携带RIOK1扩增的癌症和携带TP53突变的癌症的百分比。n=病例数,p值表示RIOK1扩增和TP53突变在同一癌症中发生的统计可能性。图A、B、C、D中的数据来自Cancer Genomics v5.4.3的cbiopportal[3]。
除了携带功能缺失突变的p53外,野生型p53在转录或蛋白质水平失调时也可能致癌(例如,错误表达、错定位到细胞质上阻碍其核活动、与p53结合的病毒致癌蛋白失活)[27]。在MDM2扩增或过表达或不能抑制MDM2的细胞中观察到p53的组成性降解[28]。如图1A所示,riok1激活的mTORC2激酶复合物和Ras GTPase-PI3K轴通过激酶Akt下调p53,从而刺激MDM2[29,30]。此外,无论是否有H/N/K-Ras和转录因子NF-κB的支持,c-Myc都能驱动Aurora A和B的表达[31,32]。虽然Aurora B可以磷酸化并稳定c-Myc[33],但这两种激酶也针对p53,要么降低其活性,要么通过MDM2刺激其泛素化[31,34](图1A)。此外,c-Myc激活甲基转移酶WDR4的表达,该酶在某些trna的46位催化7-甲基鸟苷修饰,从而刺激cyclin B1转录本的翻译。Cyclin B1促进p53泛素化和降解[35]。由c-Myc转录控制的Cyclin A2[36]也可以抑制p53和细胞凋亡[37,38],其机制尚不清楚。Cyclin A2进一步通过整合素信号传导触发EMT通路[39]。
c-Myc促进长链非编码RNA MILIP的产生[40]。通过竞争与p53结合,MILIP阻止p53被TRIML2 summoylation,从而使p53泛素化[40]。最后,长链非编码RNA LincROR会隔离miR-204-5p,从而下调RioK1和MDM2[41](图1A)。值得注意的是,c-Myc、NF-κB[18]、Ras和PI3K/Akt[42]共同诱导EMT转录因子STAT3[43]和TWIST1[44]的表达或激活,这两个转录因子都可以调节p53。的确,通过与p53相互作用,TWIST1阻止了p53稳定所需的磷酸化,从而导致p53降解[45]。STAT3结合TP53启动子抑制其转录[46]。此外,MTDH直接或通过作用于NF-κB促进PI3K/Akt活性[47],进而抑制PTEN,导致p53降解[17,48]。
RioK1可能传递给p53的上游线索包括营养可利用性。在人类和果蝇中,RioK1激活mTORC2复合物[13],该复合物发出营养物质存在的信号,以调节代谢、蛋白质合成、生长和增殖。在酿酒酵母中,同源物Rio1在营养匮乏时限制生长和分裂,而在营养丰富时促进生长和分裂。在后一种情况下,Rio1在转录水平上自动激活自身[25]。转录组、相互作用组和激酶组分析表明,酵母Rio1也可能是热、渗透和氧化应激的信号[25]。在古细菌中,通常从极端环境中分离出来,在紫外线照射[49,50,51]和伽马射线治疗[52]下,RIO1的表达和活性上调,进一步表明这种保守的激酶/ atp酶在dna损伤反应中起作用。在人类细胞中探索这些观察结果对于进一步巩固RioK1、p53和细胞存活之间的关系将是重要的。
RioK1对(癌症)细胞存活至关重要[21]。事实上,只有低比例(< 3%)的癌症在基因中携带深度缺失或突变(图2B),但由于仍未识别的遗传适应而生长和增殖。泛癌症研究[3,4,9]也显示,RIOK1的扩增频率较低(图2B, D),而其过表达在整个癌症谱系中都很突出(图2A, C)。相反,只有少数恶性肿瘤存在RIOK1过表达,其中甲状腺、前列腺和乳腺癌最为普遍(图2A)。RioK1水平的升高可能源于c-Myc的转录上调[21]和/或通过CK2或LSD1增加的蛋白质稳定性[16](图1A)。RioK1浓度升高与高肿瘤分级、肿瘤侵袭性和低患者生存率相关[10,11,15]。当RIOK1过表达时,p53降解,癌细胞对治疗变得难治[2](图1B)。例如,结直肠癌细胞对放疗的耐药[2],非小细胞肺癌细胞对顺铂的耐药[11,53],雌激素受体阳性的乳腺癌细胞对他莫昔芬的耐药[54],结直肠癌细胞对5-氟尿嘧啶的耐药[55]。p53低表达、失调或突变失活的恶性肿瘤同样会对治疗产生耐药性[56],这加强了一种假设,即RIOK1过表达癌症的化疗和放射耐药很可能是由于p53活性的丧失。只有极低比例的乳腺癌(分析的996例病例中<1%)表现出RIOK1扩增和TP53突变的显著共存(图2D)。相反,子宫内膜癌、膀胱癌、乳腺癌和非小细胞肺癌的高比例(10-26%,图2C)表明,RIOK1过表达的发生与TP53突变存在显著的正相关[3],表明后一种情况并非相互排斥。这一发现值得进一步的研究,因为它可以很好地促进我们对p53突变时RioK1水平/活性升高如何影响(癌症)细胞生物学的理解。
下调RIOK1被证明可以阻止egf驱动和ras成瘾癌症的增殖和侵袭性[9,10,13]。其他研究表明,当RioK1缺失时,mtap缺失[57,58,59]或ras驱动的恶性肿瘤[15,60,61]具有致命性。这些发现揭示了由K/H/N-Ras和构成RioK1-p53网络的其他致癌因子(如c-Myc、Akt[12]、mTOR)驱动的癌症对RioK1缺乏的遗传易感性,表明药物靶向RioK1可能是癌症临床中一种成功的策略。在进化过程中,大多数真核蛋白激酶的催化结构域在结构上保持相似或“典型”。然而,RioK1的激酶/ atp酶结构域发育不典型[5],因为它的c端叶只包含6个典型α-螺旋中的3个,而另外两个α-螺旋位于5个β-片附近,从而延长了n端叶。RioK1在αC和β3之间也有一个灵活的31个残基插入,缺乏激活环[8,62,63]。它独特的组成可以激发高选择性RioK1抑制剂的开发。Toyocamycin在67年前被发现是一种由toyocaencis链霉菌产生的抗念珠菌抗生素[64],目前在实验室环境中用于抑制RioK1[2,21,24]。由于这种化合物是ATP类似物,它可能很好地抑制其他(a)典型的蛋白激酶。不幸的是,没有这方面的数据。在最近的研究中,左西孟旦,一种用于治疗心力衰竭的腙和吡嗪衍生物,被预测以atp竞争的方式靶向RioK1[65]。事实上,许多癌症被证明对左西孟旦敏感,并对造血淋巴瘤细胞系观察到最强的抗肿瘤作用。然而,该化合物也可能靶向其他激酶,包括RioK3[65],但这方面的实验证据仍然缺乏。最近,一种被批准用于特发性肺纤维化的酪氨酸激酶抑制剂Nintedanib也靶向结直肠癌细胞中的RioK1[66]。更理想的精确方法可能包括开发靶向RioK1翻译后修饰的配体(p-S407, p-T410, met-K411),这些修饰决定了RioK1的活性和稳定性。或者,围绕其催化结构域或riok1 -底物相互作用的独特折叠也可以被探测。对于后者,最近的一项研究报道了一种大环化合物,它可以在体外消除RioK1-PRMT5相互作用[67],在人类细胞中,可以引起核糖体应激反应和p53诱导的细胞凋亡。恢复失去功能的突变体p53活性的小分子也正在开发中[68]。这些药物可与破坏MDM2-p53相互作用的化合物[56]或靶向RioK1的化合物联合使用,以增强重新激活的突变型p53的稳定性,类似于在表达野生型p53的癌细胞中抑制RioK1[2]。最近的报道显示,基因下调或药物(尼达尼布)治疗RioK1可抑制表达功能获得突变体p53的恶性肿瘤的生长和增殖[15,66]。然而,考虑到p53的稳定性和活性均未被研究,并且RioK1在细胞中介导多种作用,而尼达尼布并不专门针对RioK1,因此不能排除两项研究都特异性地影响了次级活性,从而导致了所观察到的细胞毒性。
总之,我们希望这里概述的信号网络将指导和推进RioK1, p53及其功能关系的未来研究;建立RioK1作为一种有价值的生物标志物,并在由p53突变或RioK1-p53网络失调驱动的广泛癌症中激发针对RioK1的新疗法。
ccDownload: /内容/ pdf / 10.1038 / s41420 - 023 - 01704 - 7. - pdf




